|
有機分子の機能設計
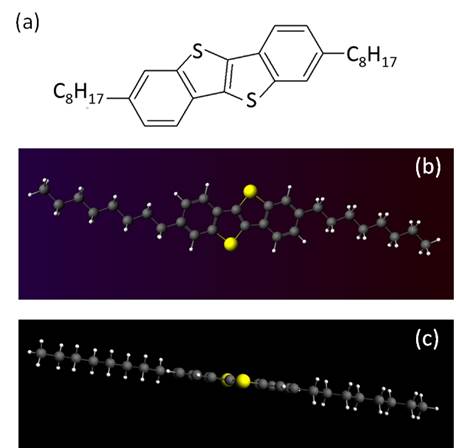
本研究室の基本テーマである「フレキシブル液晶ディスプレイ」において、その構成部材となるのは、液晶、高分子、有機半導体などの機能性有機材料です。これらの新たな電子・光機能を創出するには、それらを支配する分子軌道・分子間の軌道の重なり(相互作用)などを深く理解することが不可欠です。これらの挙動を紐解く手段として、密度汎関数法による量子化学計算を導入しながら、有機分子の機能設計を進めています。 以下に、近年高移動度な有機半導体材料として盛んに研究が行われているベンゾチエノベンゾチオフェン誘導体(C8BTBT, 図1(a))の計算結果を例示します。 量子化学計算では、分子を構成する電子の波動関数を解くことにより電子軌道を求めます。これにより分子の最安定構造、光学特性、電子物性の算出が可能になります。しかし、その厳密解を求めることは一般的に困難なため、近似を駆使した高精度な計算手法が開発されています。図1では最もよく用いられるB3LYPと呼ばれる交換相関汎関数を用いた近似計算手法により、単一分子の最安定構造の計算(構造最適化)を行った結果を示します。アルキル鎖が分子平面内でやや屈曲した構造を取った分子配座を取ると同時に、π電子共役骨格部の平面性が非常に高いことが分かります。
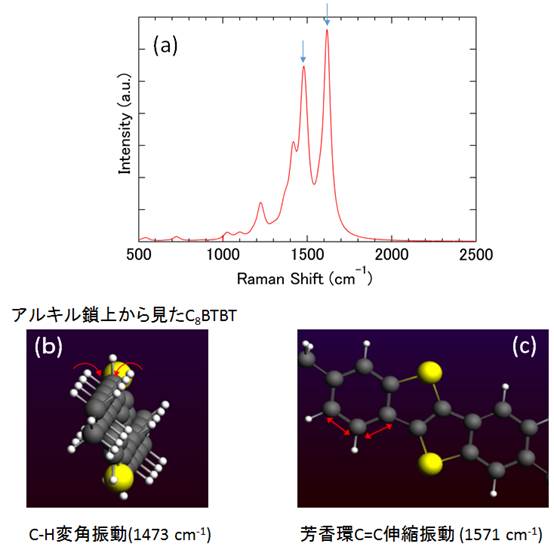
構造を最適化したC8BTBT単一分子の結果をもとに、調和振動子モデルから基準振動スペクトル(ラマンシフト)を計算した結果を図2に示します。波数に対する振動モードを予測し、ラマン散乱測定の実験結果と照合することで分子配向や残留不純物の評価等に役立てることが可能です。なお、構造最適化の結果は最安定のエネルギー状態(基底状態)に相当するものです。従って励起状態の計算により、基底状態から励起状態への遷移に対応する光吸収スペクトルの予測も可能となります。
|